Introduction
In this article, we will explore how to perform homocoupling reactions directly in the laboratory.
Specifically, we will examine standard procedures for the homocoupling of aryl halides or triflates using Cu, Ni, and Pd catalysts.

Homocoupling with Cu as catalyst-The Ullmann reaction
General reaction conditions
The homocoupling reaction of aryl halides catalyzed by copper is commonly known as the Ullmann reaction. We have discussed this transformation in our previous articles: Ullmann Coupling – The First Publication and Ullmann Coupling – An Overview. You may refer to both for a comprehensive understanding of this reaction.
In this article, however, we aim to highlight the general reaction conditions typically employed for the Ullmann homocoupling reaction.
Specifically, the Ullmann reaction is usually carried out using:
Stoichiometric amounts of metallic copper (Cu⁰);
Elevated temperatures (generally above 200 °C);
Aryl iodides and aryl bromides as the most common substrates.
The classic Ullmann reaction was originally performed without any solvent, simply by heating copper powder with the aryl halide at the boiling point of the halide until the reaction reached completion. More modern approaches, however, recommend the use of solvents such as DMF. In fact, it is now widely accepted that DMF can facilitate the Ullmann coupling at lower temperatures and with reduced catalyst loading.
Several modifications have been proposed to further improve the efficiency of the reaction, including:
The use of activated copper powder—specifically, freshly prepared copper from CuI and potassium—which allows the reaction to proceed at significantly lower temperatures (around 85 °C);
The application of ultrasound irradiation, which can notably accelerate the reaction rate;
Regarding aryl halide reactivity, substrates bearing electron-withdrawing groups in the ortho position relative to the halogen are generally more reactive. Conversely, aryl halides substituted with functional groups such as –OH, –NH₂, or –COOH can interfere with the reaction by introducing additional coordination sites for copper, thus limiting the efficiency of the coupling.
In the following section, typical procedures for Ullmann homocoupling are described in detail.
Typical procedures with Cu
After discussing the general characteristics of Ullmann trasformation, let’s finally see how to carry out the reaction in the laboratory. Below are some procedures found in the literature.
Classic Ullmann reaction
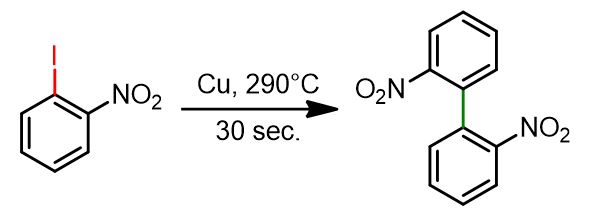
2-Iodonitrobenzene (249 mg, 1.0 mmol) was placed in a 15 cm test tube along with copper powder (191 mg, 3.0 mmol) and sand (200 mg). The resulting mixture was heated in a sand bath at 350 °C, above the boiling point of 2-iodonitrobenzene (approximately 290 °C), for 20–30 seconds. As the reaction proceeded, 2-iodobenzonitrile melted and began to boil. After cooling, the dry residue was subjected to column chromatography for purification, using a gradient from dichloromethane:hexane (30:70) to dichloromethane:ethyl acetate (90:10). The final product was obtained in 80–90% yield. [1]
Ullmann reaction with Cu in DMF
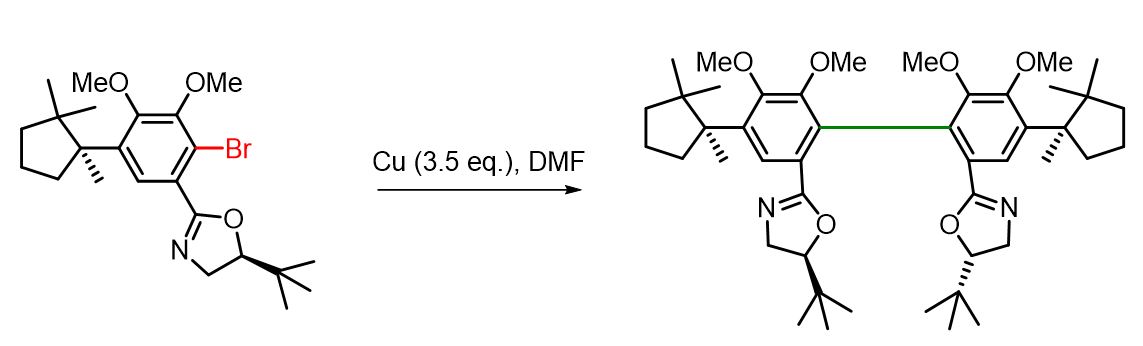
(S)-tert-Leucinol-o-bromoaryloxazoline (300 mg, 0.66 mmol) was placed in a 10 mL flask along with freshly activated copper powder (150 mg, 2.3 mmol) and DMF (1 mL). The mixture was stirred and heated at 95 °C for 8 hours. It was then diluted with an additional 5 mL of DMF, and a reflux condenser was attached to the apparatus. The reaction mixture was further heated under reflux for 3 days. After cooling, the mixture was filtered through cotton to remove the excess copper. The filtrate was poured into water and extracted with diethyl ether (3×). The combined organic layers were washed with water (2×) and brine, dried over anhydrous Na₂SO₄, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (50% EtOAc/Hex), affording the biaryl–copper complex. This complex was dissolved in ether and washed with aqueous NH₄OH (2×) and brine. The organic layers were dried over Na₂SO₄ and concentrated under reduced pressure to yield the desired product as a single atropisomer in 64% yield (158 mg).[2]
Homocoupling with Ni as catalyst
General reaction conditions
The earliest publications on the use of nickel in homocoupling reactions reported the application of stoichiometric amounts of Ni(0) in the presence of a ligand. The reaction shown below serves as a representative example.
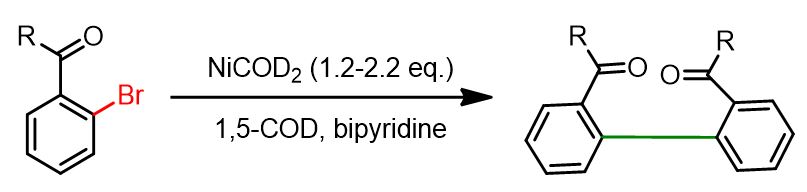
However, this approach presented two main drawbacks: the requirement for stoichiometric amounts of nickel and the need to operate in a glovebox, as Ni(0) catalysts are generally unstable under aerobic conditions.
To address these limitations, alternative strategies were developed—most notably, the use of Ni(II) precursors, which are reduced in situ to the active Ni(0) species. This advancement eliminated the need for a glovebox. However, it still required stoichiometric amounts of Ni(II). Among various reductants tested, zinc powder proved to be the most effective. See the reaction below for a representative example.
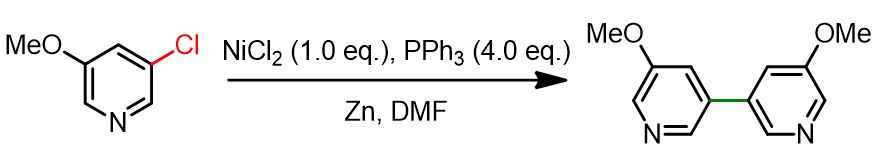
Nowadays, nickel-catalyzed homocoupling reactions can be performed using catalytic amounts of Ni(II) species, under the following general conditions:
A reducing agent to convert Ni(II) into the active Ni(0) form. Zinc powder is the most commonly used reductant, although other systems—such as LiH in the presence of a base like t-BuOLi—have also been reported;
Mild reaction temperatures, typically in the range of 50–100 °C.
When zinc is used as the reducing agent, iodide salts are often added as additives, as iodide ions are believed to accelerate the reaction.
In the following section, typical procedures for Ni-catalyzed homocoupling reactions are presented, providing valuable insights into how these reactions can be practically carried out in the laboratory.
Typical procedures with Ni
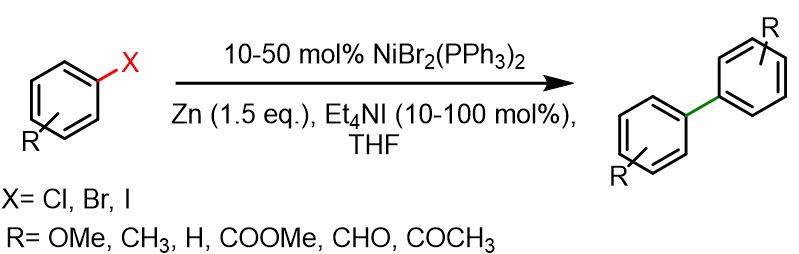
In a two-necked 50 ml round bottom flask, NiBr2(PPh3)2 (0.5-2.5 mmol), Zn dust (7.5 mmol) and Et4NI (0.5-5 mmol) were inserted under Ar atmosphere. Then dry THF was added and the mixture was stirred for 30 min at room temperature. Afterwards a solution of aryl halide (5 mmol) in THF (5 ml), previously purged with Ar, was added in the mixture which was stirred for 4-20 h at 50°C. Filtration was performed to separate the inorganic precipitation followed by washing with benzene; the combined organic layers were evaporated under reduced pressure and the obtained residue was purified with column chromatography on silica gel (50 g). Typical yields of biaryl were in the range 57 – 99%.[3]
In the following reaction, the Ni catalyst promotes the homocoupling of aryl triflates in the presence of zinc powder as a reducing agent. The reaction proceeds more efficiently with electron-donating groups than with electron-withdrawing ones. Aryl triflates bearing CN or Cl substituents tend to form byproducts due to the activation of the corresponding functional groups (CN and Cl groups respectively).
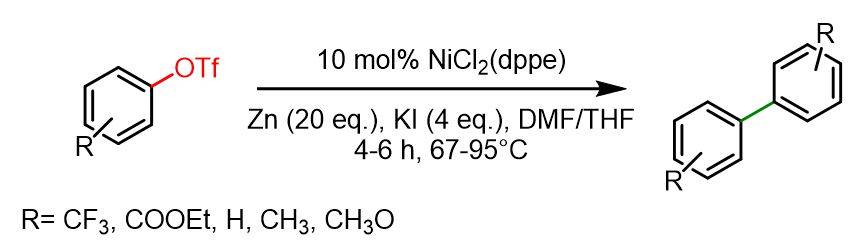
Dry THF and dry DMF (1 ml + 1ml) were added to a mixture of activated zinc powder (3.95 g, 60.0 mmol), KI (2g, 12 mmol) and NiCl2(dppe) (0.158 g, 0.3 mmol). Then p-R-aryl triflate (3 mmol) was added. The reaction mixture was refluxed at 67°C or 95°C for 4-6 h. Afterwards it was hydrolized with saturated solution of NH4Cl and extracted with diethyl ether. Where necessary, column chromatography on silica gel (petroleum ether/ethyl acetate as eluent) was performed. The yields of biaryls were in the range 86-99%.[4]
Homocoupling with Pd as catalyst
General reaction conditions
Palladium-catalyzed homocoupling reactions have emerged as a valuable alternative to their copper and nickel counterparts. However, specific conditions must be met for successful implementation of Pd-catalyzed homocoupling, including:
The use of a reducing agent capable of regenerating the active Pd(0) species from Pd(II) intermediates formed during the reaction.
The presence of a reductant is essential to allow the reaction to proceed with catalytic amounts of palladium. To better understand the importance of this reductant, it is useful to examine the reaction mechanism in detail.[5]
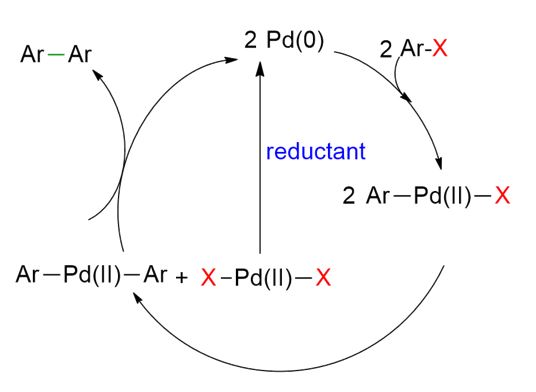
As shown in the figure above, the Pd-catalyzed homocoupling reaction of aryl halides begins with the oxidative addition of Ar–X to Pd(0), forming an Ar–Pd(II)–X intermediate. It is generally proposed that two molecules of Ar–Pd(II)–X undergo a ligand exchange, generating two new species: Ar–Pd(II)–Ar and X–Pd(II)–X.
The Ar–Pd(II)–Ar species then undergoes reductive elimination to yield the desired biaryl product (Ar–Ar) and regenerate Pd(0), thus continuing the catalytic cycle. In contrast, the X–Pd(II)–X species cannot re-enter the catalytic cycle unless it is reduced back to Pd(0) by an external reducing agent.
This highlights why a reductant is essential in Pd-catalyzed homocoupling reactions: without it, a significant portion of the palladium remains trapped as PdX₂, which is catalytically inactive unless converted back to Pd(0).
The typical reducing agents used in Pd-catalyzed homocoupling reactions are summarized in the table below.

Typical procedures with Pd
The followong reaction is compatible with most functional groups, except for haloarenes bearing cyanide, hydroxy, carboxy, or amino substituents. Furthermore, it generally does not proceed with ortho-substituted haloarenes. Common side reactions include the dehalogenation of bromoarenes (typically 20–30%) and transarylation when AsPh₃ is used.
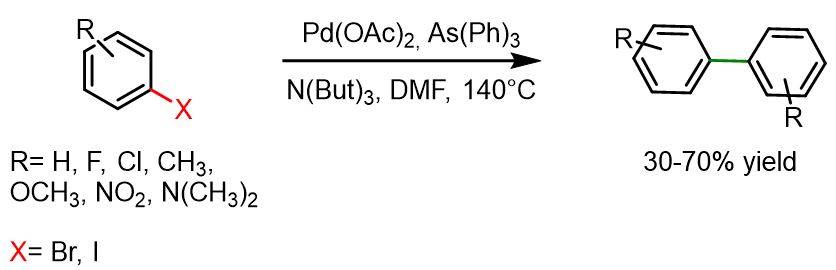
Procedure for bromoarenes: A 100 ml dried flask was filled with arylhalide (100 mmol), DMF (40 ml), N(But)3 (26 ml, 110 mmol), Pd(OAc)2 (0.112 g, 0.5 mmol), and As(Ph)3 (0.30 g, 1.0 mmol) under argon atmosphere. The reaction mixture was first degassed and purged with argon, then stirred at 140°C for 24 h. Afterwards the reaction mixture was poured into a 10% HCl aqueous solution (500 ml). Solid compounds precipitated from the mixture and were therefore isolated by filtration, followed by purification via recrystallization from ethanol. Liquid products, on the other hand, were extracted with diethyl ether and purified by distillation.
Procedure from iodoarenes: Iodoarenes were reacted following the same procedure as for bromoarenes, but without the use of triphenylarsine.[6]
The following reaction, developed by Lemaire’s team, performs well with various types of haloarenes — including those with meta, ortho, or para substitutions — as well as with 2-halopyridines and 2- or 3-haloquinolines.
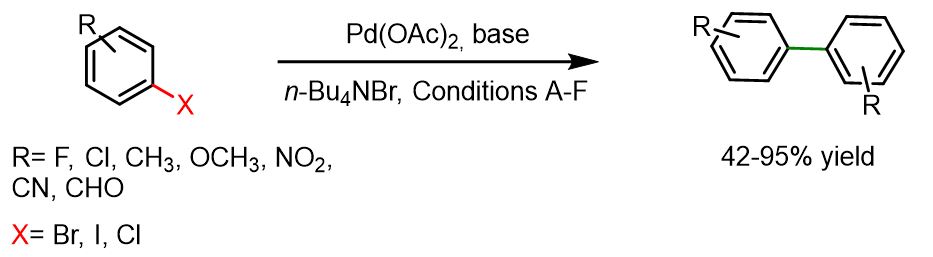
A mixture of base (8 mmol), Pd(OAc)₂ (0.4 mmol, 5 mol%), Bu₄NBr (4 mmol), and aryl halide (8 mmol) in a solvent (1.25 mL) — either toluene or a DMF/H₂O mixture (0.90/0.35 mL) — was stirred under a nitrogen atmosphere for a few minutes at 105 °C (in toluene) or 115 °C (in DMF). When required, isopropanol (8 mmol) was added, and the reaction mixture was then heated for a period ranging from 7 to 177 hours, depending on the substrate used. After cooling to room temperature, water and diethyl ether were added. The organic layer was washed with water, dried over MgSO₄, filtered, and concentrated under reduced pressure. The crude product was purified by either preparative TLC or recrystallization.[7]
The table below summarizes Conditions A–F along with the corresponding substrates to which each set of conditions was applied.

References
[1] Habgood, L. G.; Gregor, R. W. 7.1. A Solvent-Free Ullmann Coupling: Synthesis of 2,2′-Dinitrobiphenyl. In Comprehensive Organic Chemistry Experiments for the Laboratory Classroom; Afonso, C. A. M., Candeias, N. R., Simão, D. P., Trindade, A. F., Coelho, J. A. S., Tan, B., Franzén, R., Eds.; The Royal Society of Chemistry, 2016; pp 550–553. https://doi.org/10.1039/9781849739634-00550
[2] Degnan, A. P.; Meyers, A. I. Total Syntheses of (−)-Herbertenediol, (−)-Mastigophorene A, and (−)-Mastigophorene B. Combined Utility of Chiral Bicyclic Lactams and Chiral Aryl Oxazolines. J. Am. Chem. Soc. 1999, 121 (12), 2762–2769. https://doi.org/10.1021/ja984182x.
[3] Iyoda, M.; Otsuka, H.; Sato, K.; Nisato, N.; Oda, M. Homocoupling of Aryl Halides Using Nickel(II) Complex and Zinc in the Presence of Et4NI. An Efficient Method for the Synthesis of Biaryls and Bipyridines. Bulletin of the Chemical Society of Japan 1990, 63 (1), 80–87. https://doi.org/10.1246/bcsj.63.80.
[4] Jutand, A.; Mosleh, A. Palladium and Nickel Catalyzed Synthesis of Biaryls from Aryl Triflates in the Presence of Zinc Powder. Synlett 1993, 1993 (08), 568–570. https://doi.org/10.1055/s-1993-22531
[5] Kotora, M.; Takahashi, T. Palladium-Catalyzed Homocoupling of Organic Electrophiles or Organometals. In Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi, E., Ed.; John Wiley & Sons, Inc.: New York, USA, 2002; pp 973–993. https://doi.org/10.1002/0471212466.ch40.
[6] Brenda, M.; Knebelkamp, A.; Greiner, A.; Heitz, W. Novel Palladium-Catalyzed Biaryl Synthesis with Haloarenes. Synlett 1991, 1991 (11), 809–810. https://doi.org/10.1055/s-1991-20885.
[7] Hassan, J.; Penalva, V.; Lavenot, L.; Gozzi, C.; Lemaire, M. Catalytic Alternative of the Ullmann Reaction. Tetrahedron 1998, 54 (45), 13793–13804. https://doi.org/10.1016/S0040-4020(98)00849-7
2 replies on “Homocoupling reactions of aryl halides-Typical procedures with Cu, Ni and Pd catalysts”
Nice share !
Thank you!